Lecture 23 — Machine Learning IV#
Graph Neural Networks for Physics#
Why Graphs?#
Many physical systems are naturally described as graphs — a collection of nodes connected by edges:
Physical system |
Nodes |
Edges |
|---|---|---|
Molecule |
Atoms |
Bonds / distance cutoff |
Crystal |
Atoms in unit cell |
Nearest neighbours |
Protein |
Amino acid residues |
Spatial proximity |
Social network |
People |
Friendships |
Electrical circuit |
Components |
Wires |
The key insight: the structure of the system matters. An MLP treats inputs as a flat vector and ignores which atoms are close to which. A GNN respects the connectivity.
From Lecture 23: we built permutation-invariant networks using Deep Sets (process each element, then sum). A GNN generalises this by also considering which elements are connected.
import numpy as np
import matplotlib.pyplot as plt
import torch
import torch.nn as nn
import torch.nn.functional as F
plt.rcParams['figure.figsize'] = [6, 4]
plt.rcParams['font.size'] = 9
print(f"PyTorch {torch.__version__}")
PyTorch 2.6.0
I. Graphs: The Data Structure#
A graph \(\mathbf{G} = (\mathbf{V}, \mathbf{E})\) consists of:
Nodes \(\mathbf{V} = \{v_1, \ldots, v_N\}\) with features \(\mathbf{h}_i \in \mathbb{R}^d\)
Edges \(\mathbf{E} = \{(i, j)\}\) with optional features \(\mathbf{e}_{ij} \in \mathbb{R}^k\)
Neighbours of node \(i\): \(\mathbf{N}(i) = \{j : (i,j) \in \mathbf{E}\}\)
For molecular systems:
Node features: Atomic number, element type (one-hot encoded)
Edge features: Distance \(r_{ij}\), bond type
Connectivity: All atom pairs within a cutoff distance \(r_c\)
# Example: represent a water molecule as a graph
# H2O: O at center, two H atoms
# Atom positions (Angstroms)
positions = np.array([
[0.000, 0.000, 0.000], # O
[0.757, 0.587, 0.000], # H
[-0.757, 0.587, 0.000], # H
])
atomic_numbers = [8, 1, 1] # O, H, H
elements = ['O', 'H', 'H']
# Build graph: connect atoms within cutoff
cutoff = 2.0 # Angstroms
edges = []
edge_distances = []
for i in range(len(positions)):
for j in range(len(positions)):
if i != j:
d = np.linalg.norm(positions[i] - positions[j])
if d < cutoff:
edges.append([i, j])
edge_distances.append(d)
edges = np.array(edges)
edge_distances = np.array(edge_distances)
print("Water molecule graph:")
print(f" Nodes: {len(positions)} atoms ({elements})")
print(f" Edges: {len(edges)} connections")
for e, d in zip(edges, edge_distances):
print(f" {elements[e[0]]} ({e[0]}) -- {elements[e[1]]} ({e[1]}): {d:.3f} A")
Water molecule graph:
Nodes: 3 atoms (['O', 'H', 'H'])
Edges: 6 connections
O (0) -- H (1): 0.958 A
O (0) -- H (2): 0.958 A
H (1) -- O (0): 0.958 A
H (1) -- H (2): 1.514 A
H (2) -- O (0): 0.958 A
H (2) -- H (1): 1.514 A
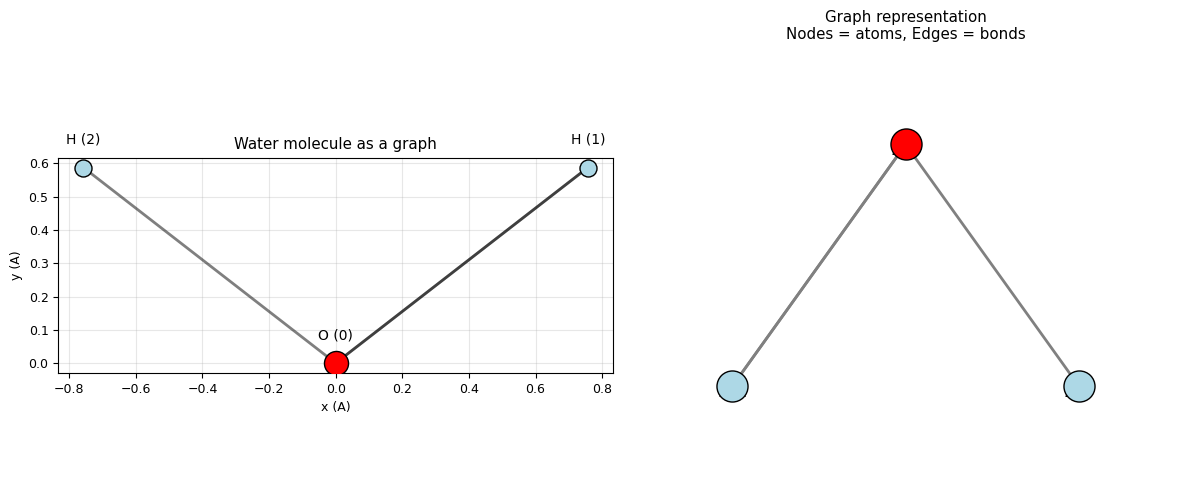
# Visualise the molecular graph
fig, axes = plt.subplots(1, 2, figsize=(12, 5))
# 2D projection of molecule
ax = axes[0]
colors_atom = {'O': 'red', 'H': 'lightblue'}
sizes_atom = {'O': 300, 'H': 150}
for e in edges[:len(edges)//2]: # undirected, so plot each pair once
p1, p2 = positions[e[0]], positions[e[1]]
ax.plot([p1[0], p2[0]], [p1[1], p2[1]], 'k-', lw=2, alpha=0.5)
for i, (pos, elem) in enumerate(zip(positions, elements)):
ax.scatter(pos[0], pos[1], c=colors_atom[elem], s=sizes_atom[elem],
edgecolors='black', zorder=5)
ax.annotate(f'{elem} ({i})', (pos[0], pos[1]), fontsize=10,
ha='center', va='bottom', xytext=(0, 15),
textcoords='offset points')
ax.set_xlabel('x (A)'); ax.set_ylabel('y (A)')
ax.set_title('Water molecule as a graph')
ax.set_aspect('equal')
ax.grid(alpha=0.3)
# Show the graph representation schematically
ax2 = axes[1]
# Draw abstract graph
node_pos = {0: (0.5, 1), 1: (0, 0), 2: (1, 0)}
for e in edges[:len(edges)//2]:
p1, p2 = node_pos[e[0]], node_pos[e[1]]
ax2.annotate('', xy=p2, xytext=p1,
arrowprops=dict(arrowstyle='<->', color='gray', lw=2))
for i, elem in enumerate(elements):
pos = node_pos[i]
ax2.scatter(*pos, c=colors_atom[elem], s=500, edgecolors='black', zorder=5)
ax2.text(pos[0], pos[1], f'{elem}\nZ={atomic_numbers[i]}',
ha='center', va='center', fontsize=9, fontweight='bold')
ax2.set_xlim(-0.3, 1.3); ax2.set_ylim(-0.4, 1.4)
ax2.set_title('Graph representation\nNodes = atoms, Edges = bonds')
ax2.axis('off')
plt.tight_layout()
plt.show()
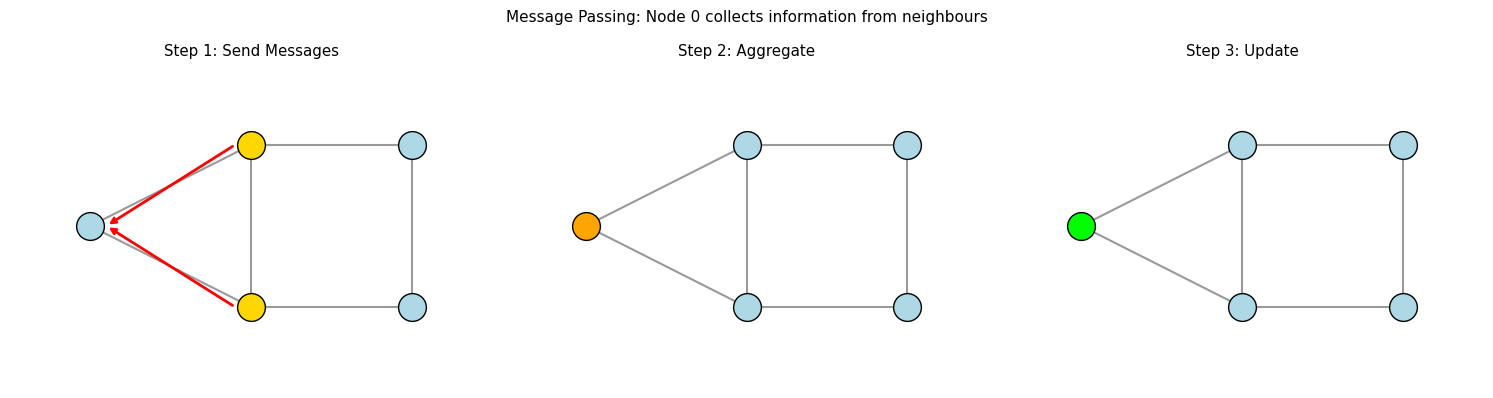
II. Message Passing: How GNNs Work#
The core operation in a GNN is message passing. In each layer:
Message: Each node sends a message to its neighbours based on its current features
Aggregate: Each node collects messages from all its neighbours
Update: Each node updates its features based on the aggregated messages
Formally, one message-passing layer updates node \(i\)’s features as:
where:
\(\psi\) = message function (what information to send)
\(\bigoplus\) = aggregation (sum, mean, or max over neighbours)
\(\phi\) = update function (how to combine old features with messages)
After \(L\) layers, each node’s features contain information about its \(L\)-hop neighbourhood — this is the GNN’s receptive field.
# Visualise message passing
fig, axes = plt.subplots(1, 3, figsize=(15, 4))
# Simple 5-node graph
node_pos = {0: (0, 0), 1: (1, 0.5), 2: (1, -0.5), 3: (2, 0.5), 4: (2, -0.5)}
edges_vis = [(0, 1), (0, 2), (1, 3), (2, 4), (1, 2), (3, 4)]
titles = ['Step 1: Send Messages', 'Step 2: Aggregate', 'Step 3: Update']
highlights = [
{1: 'gold', 2: 'gold'}, # messages from neighbours
{0: 'orange'}, # aggregation at node 0
{0: 'lime'}, # updated node
]
for ax, title, hl in zip(axes, titles, highlights):
# Draw edges
for (i, j) in edges_vis:
p1, p2 = node_pos[i], node_pos[j]
ax.plot([p1[0], p2[0]], [p1[1], p2[1]], 'k-', lw=1.5, alpha=0.4)
# Draw nodes
for i, pos in node_pos.items():
color = hl.get(i, 'lightblue')
ax.scatter(*pos, s=400, c=color, edgecolors='black', zorder=5)
ax.text(pos[0], pos[1], str(i), ha='center', va='center', fontsize=11)
ax.set_title(title)
ax.set_xlim(-0.5, 2.5); ax.set_ylim(-1, 1)
ax.axis('off')
# Add arrows for messages in step 1
for i in [1, 2]:
p1, p2 = node_pos[i], node_pos[0]
axes[0].annotate('', xy=(p2[0]+0.1, p2[1]), xytext=(p1[0]-0.1, p1[1]),
arrowprops=dict(arrowstyle='->', color='red', lw=2))
plt.suptitle('Message Passing: Node 0 collects information from neighbours', fontsize=11)
plt.tight_layout()
plt.show()
III. Building a GNN from Scratch#
Let’s implement a simple message-passing GNN in pure PyTorch.
Our GNN will predict the total energy of a system of Lennard-Jones atoms from their positions and types.
class MessagePassingLayer(nn.Module):
"""
One layer of message passing.
For each edge (i, j):
message_ij = MLP([h_i, h_j, e_ij])
For each node i:
aggregated_i = sum of messages from neighbours
h_i_new = MLP([h_i, aggregated_i])
"""
def __init__(self, node_dim, edge_dim, hidden_dim):
super().__init__()
# Message function: combines sender, receiver, and edge features
self.message_mlp = nn.Sequential(
nn.Linear(2 * node_dim + edge_dim, hidden_dim),
nn.SiLU(),
nn.Linear(hidden_dim, hidden_dim),
)
# Update function: combines old features with aggregated messages
self.update_mlp = nn.Sequential(
nn.Linear(node_dim + hidden_dim, hidden_dim),
nn.SiLU(),
nn.Linear(hidden_dim, node_dim),
)
def forward(self, h, edge_index, edge_attr):
"""
h: (N, node_dim) node features
edge_index: (2, E) source and target node indices
edge_attr: (E, edge_dim) edge features
"""
src, dst = edge_index # source and destination nodes
N = h.size(0)
# 1. Compute messages
msg_input = torch.cat([h[src], h[dst], edge_attr], dim=1)
messages = self.message_mlp(msg_input) # (E, hidden_dim)
# 2. Aggregate messages (sum for each target node)
agg = torch.zeros(N, messages.size(1), device=h.device)
agg.index_add_(0, dst, messages) # sum messages by target
# 3. Update node features
update_input = torch.cat([h, agg], dim=1)
h_new = h + self.update_mlp(update_input) # residual connection
return h_new
print("MessagePassingLayer defined.")
print("Key operations: message (per edge) → aggregate (per node) → update (per node)")
MessagePassingLayer defined.
Key operations: message (per edge) → aggregate (per node) → update (per node)
class SimpleGNN(nn.Module):
"""
Complete GNN for predicting a scalar property (like energy)
from atomic positions.
"""
def __init__(self, n_elements=3, node_dim=32, edge_dim=16, n_layers=3):
super().__init__()
# Embed atomic number into a learnable vector
self.atom_embedding = nn.Embedding(n_elements + 1, node_dim)
# Radial basis functions to encode distances
self.n_rbf = edge_dim
self.rbf_centers = nn.Parameter(
torch.linspace(0.5, 5.0, edge_dim), requires_grad=False
)
self.rbf_width = 0.5
# Message passing layers
self.layers = nn.ModuleList([
MessagePassingLayer(node_dim, edge_dim, node_dim)
for _ in range(n_layers)
])
# Output: per-atom energy, then sum
self.output_mlp = nn.Sequential(
nn.Linear(node_dim, node_dim),
nn.SiLU(),
nn.Linear(node_dim, 1),
)
def radial_basis(self, distances):
"""Expand distances into radial basis functions (Gaussian)."""
return torch.exp(-(distances.unsqueeze(-1) - self.rbf_centers)**2
/ (2 * self.rbf_width**2))
def forward(self, z, positions, edge_index, batch=None):
"""
z: (N,) atomic numbers
positions: (N, 3) coordinates
edge_index: (2, E) graph connectivity
batch: (N,) which graph each atom belongs to (for batching)
"""
# Initial node features from atom type
h = self.atom_embedding(z) # (N, node_dim)
# Compute edge features from distances
src, dst = edge_index
diff = positions[dst] - positions[src] # (E, 3)
distances = torch.norm(diff, dim=1) # (E,)
edge_attr = self.radial_basis(distances) # (E, n_rbf)
# Message passing
for layer in self.layers:
h = layer(h, edge_index, edge_attr)
# Per-atom energy
atom_energy = self.output_mlp(h).squeeze(-1) # (N,)
# Sum per-atom energies to get total energy per molecule
if batch is None:
return atom_energy.sum()
else:
# Scatter-add by graph index
n_graphs = batch.max().item() + 1
energy = torch.zeros(n_graphs, device=h.device)
energy.index_add_(0, batch, atom_energy)
return energy
gnn = SimpleGNN()
n_params = sum(p.numel() for p in gnn.parameters())
print(f"SimpleGNN: {n_params} parameters")
print(f"Architecture: atom embedding → {len(gnn.layers)} message-passing layers → per-atom MLP → sum")
SimpleGNN: 21585 parameters
Architecture: atom embedding → 3 message-passing layers → per-atom MLP → sum
Radial Basis Functions#
Distances are continuous, but neural networks work best with expanded representations. We use Gaussian radial basis functions (RBFs):
This turns a single distance into a vector of \(K\) features, each “tuned” to a different distance range.
# Visualise radial basis functions
r = torch.linspace(0, 6, 200)
centers = torch.linspace(0.5, 5.0, 16)
width = 0.5
rbf_values = torch.exp(-(r.unsqueeze(-1) - centers)**2 / (2 * width**2))
plt.figure(figsize=(8, 4))
for i in range(16):
plt.plot(r.numpy(), rbf_values[:, i].numpy(), alpha=0.7)
plt.xlabel('Distance r (A)')
plt.ylabel('RBF value')
plt.title('Gaussian Radial Basis Functions')
plt.grid(alpha=0.3)
plt.tight_layout()
plt.show()
print(f"A distance r = 2.5 A is encoded as a {len(centers)}-dim vector:")
r_test = torch.tensor([2.5])
encoded = torch.exp(-(r_test.unsqueeze(-1) - centers)**2 / (2 * width**2))
print(f" {encoded.numpy().round(3).flatten()}")
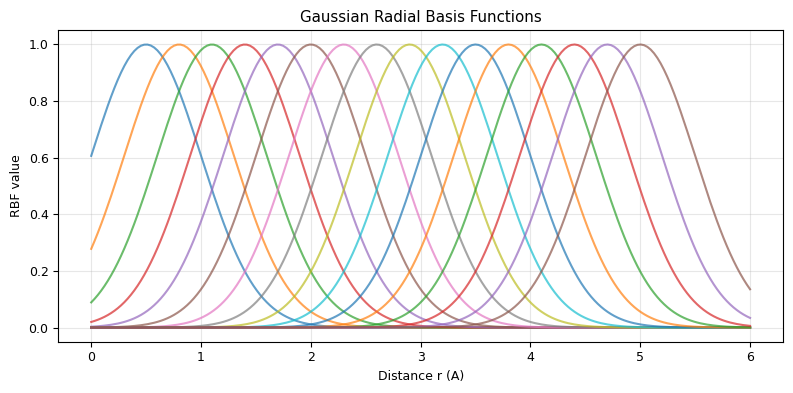
A distance r = 2.5 A is encoded as a 16-dim vector:
[0. 0.003 0.02 0.089 0.278 0.607 0.923 0.98 0.726 0.375 0.135 0.034
0.006 0.001 0. 0. ]
IV. Training the GNN: Lennard-Jones Clusters#
Let’s train our GNN to predict the total energy of small LJ clusters (continuing from Lecture 22).
def lj_energy(positions, epsilon=1.0, sigma=1.0):
"""Total LJ energy for a cluster."""
N = len(positions)
E = 0.0
for i in range(N):
for j in range(i+1, N):
r = np.linalg.norm(positions[i] - positions[j])
sr6 = (sigma / r) ** 6
E += 4 * epsilon * (sr6**2 - sr6)
return E
def build_graph(positions, cutoff=3.5):
"""Build edge list from positions within cutoff."""
N = len(positions)
src, dst = [], []
for i in range(N):
for j in range(N):
if i != j:
r = np.linalg.norm(positions[i] - positions[j])
if r < cutoff:
src.append(i)
dst.append(j)
return np.array([src, dst])
# Generate dataset of 4-atom LJ clusters
np.random.seed(42)
n_samples = 2000
n_atoms = 4
dataset = []
for _ in range(n_samples * 2): # generate extra, filter bad ones
pos = np.random.randn(n_atoms, 3) * 0.8
# Check minimum distance
dists = []
for i in range(n_atoms):
for j in range(i+1, n_atoms):
dists.append(np.linalg.norm(pos[i] - pos[j]))
if min(dists) < 0.8:
continue
E = lj_energy(pos)
edge_index = build_graph(pos)
dataset.append({
'positions': pos.astype(np.float32),
'z': np.ones(n_atoms, dtype=np.int64), # all same atom type
'energy': E,
'edge_index': edge_index.astype(np.int64),
})
if len(dataset) >= n_samples:
break
print(f"Generated {len(dataset)} valid clusters")
print(f"Energy range: [{min(d['energy'] for d in dataset):.2f}, {max(d['energy'] for d in dataset):.2f}]")
Generated 2000 valid clusters
Energy range: [-4.36, 77.49]
# Simple batching for GNNs
# Key trick: combine multiple graphs into one big graph with a `batch` vector
def collate_graphs(graph_list):
"""Batch multiple graphs into one big graph."""
all_z = []
all_pos = []
all_edges = []
all_energy = []
all_batch = []
node_offset = 0
for i, g in enumerate(graph_list):
N = len(g['z'])
all_z.append(torch.tensor(g['z']))
all_pos.append(torch.tensor(g['positions']))
all_edges.append(torch.tensor(g['edge_index']) + node_offset)
all_energy.append(g['energy'])
all_batch.append(torch.full((N,), i, dtype=torch.long))
node_offset += N
return {
'z': torch.cat(all_z),
'positions': torch.cat(all_pos),
'edge_index': torch.cat(all_edges, dim=1),
'energy': torch.tensor(all_energy, dtype=torch.float32),
'batch': torch.cat(all_batch),
}
# Quick test
test_batch = collate_graphs(dataset[:3])
print("Batched graph:")
print(f" Total nodes: {test_batch['z'].size(0)} (3 molecules x {n_atoms} atoms)")
print(f" Total edges: {test_batch['edge_index'].size(1)}")
print(f" Batch vector: {test_batch['batch']}")
print(f" Energies: {test_batch['energy']}")
Batched graph:
Total nodes: 12 (3 molecules x 4 atoms)
Total edges: 36
Batch vector: tensor([0, 0, 0, 0, 1, 1, 1, 1, 2, 2, 2, 2])
Energies: tensor([56.1277, -0.7629, -1.3259])
# Train the GNN
torch.manual_seed(42)
# Split data
n_train = int(0.8 * len(dataset))
train_data = dataset[:n_train]
test_data = dataset[n_train:]
# Normalise energies
E_mean = np.mean([d['energy'] for d in train_data])
E_std = np.std([d['energy'] for d in train_data])
for d in dataset:
d['energy_norm'] = (d['energy'] - E_mean) / E_std
# Model
gnn = SimpleGNN(n_elements=3, node_dim=32, edge_dim=16, n_layers=3)
optimizer = torch.optim.Adam(gnn.parameters(), lr=0.002)
scheduler = torch.optim.lr_scheduler.ReduceLROnPlateau(optimizer, patience=20, factor=0.5)
batch_size = 32
train_losses, val_losses = [], []
for epoch in range(200):
gnn.train()
np.random.shuffle(train_data)
epoch_loss = 0
n_batches = 0
for i in range(0, len(train_data), batch_size):
batch = collate_graphs(train_data[i:i+batch_size])
optimizer.zero_grad()
E_pred = gnn(batch['z'], batch['positions'], batch['edge_index'], batch['batch'])
E_true = torch.tensor([d['energy_norm'] for d in train_data[i:i+batch_size]],
dtype=torch.float32)
loss = F.mse_loss(E_pred, E_true)
loss.backward()
optimizer.step()
epoch_loss += loss.item()
n_batches += 1
train_losses.append(epoch_loss / n_batches)
# Validation
gnn.eval()
with torch.no_grad():
val_batch = collate_graphs(test_data)
E_pred_val = gnn(val_batch['z'], val_batch['positions'],
val_batch['edge_index'], val_batch['batch'])
E_true_val = torch.tensor([d['energy_norm'] for d in test_data],
dtype=torch.float32)
val_loss = F.mse_loss(E_pred_val, E_true_val).item()
val_losses.append(val_loss)
scheduler.step(val_loss)
if epoch % 40 == 0:
print(f"Epoch {epoch:3d} Train: {train_losses[-1]:.5f} Val: {val_loss:.5f}")
print(f"\nFinal val loss: {val_losses[-1]:.5f}")
Epoch 0 Train: 1.35160 Val: 1.02464
Epoch 40 Train: 0.04884 Val: 0.03346
Epoch 80 Train: 0.03977 Val: 0.02687
Epoch 120 Train: 0.01684 Val: 0.01145
Epoch 160 Train: 0.02407 Val: 0.01994
Final val loss: 0.01001
# Evaluate: parity plot
gnn.eval()
with torch.no_grad():
val_batch = collate_graphs(test_data)
E_pred = gnn(val_batch['z'], val_batch['positions'],
val_batch['edge_index'], val_batch['batch']).numpy()
# Un-normalise
E_pred_real = E_pred * E_std + E_mean
E_true_real = np.array([d['energy'] for d in test_data])
fig, axes = plt.subplots(1, 3, figsize=(15, 4))
# Parity plot
axes[0].scatter(E_true_real, E_pred_real, s=10, alpha=0.5)
lims = [min(E_true_real.min(), E_pred_real.min()),
max(E_true_real.max(), E_pred_real.max())]
axes[0].plot(lims, lims, 'r--', lw=2)
axes[0].set_xlabel('True Energy'); axes[0].set_ylabel('GNN Energy')
axes[0].set_title('Parity Plot'); axes[0].grid(alpha=0.3)
# Error distribution
errors = E_pred_real - E_true_real
axes[1].hist(errors, bins=40, edgecolor='black', alpha=0.7)
axes[1].set_xlabel('Error'); axes[1].set_ylabel('Count')
axes[1].set_title(f'MAE = {np.abs(errors).mean():.4f}')
axes[1].axvline(0, color='red', ls='--')
# Training curve
axes[2].semilogy(train_losses, 'b-', alpha=0.7, label='Train')
axes[2].semilogy(val_losses, 'r-', alpha=0.7, label='Val')
axes[2].set_xlabel('Epoch'); axes[2].set_ylabel('MSE')
axes[2].set_title('Training progress')
axes[2].legend(); axes[2].grid(alpha=0.3)
plt.tight_layout()
plt.show()
from sklearn.metrics import r2_score
print(f"R² = {r2_score(E_true_real, E_pred_real):.4f}")
print(f"MAE = {np.abs(errors).mean():.4f} epsilon")
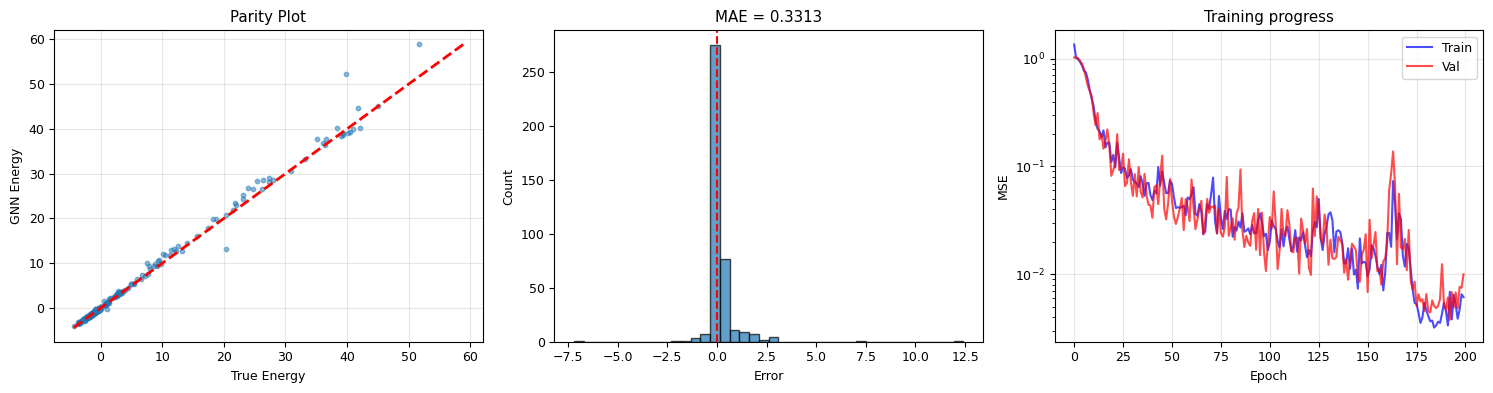
R² = 0.9902
MAE = 0.3313 epsilon
Why GNN over MLP?#
Property |
MLP (Lecture 22) |
GNN |
|---|---|---|
Input |
Fixed-size vector |
Variable-size graph |
Permutation invariance |
Must sort features |
Built in (sum aggregation) |
Scalability |
Fixed \(N\) atoms |
Any \(N\) atoms (same model!) |
Locality |
All-to-all |
Only neighbours interact |
Interpretability |
Black box |
Per-atom contributions |
V. Equivariant GNNs: Encoding Rotational Symmetry#
Our simple GNN uses distances as edge features — distances are rotation-invariant, so the predicted energy is rotation-invariant. Good!
But what about forces? Forces are vectors that rotate with the system. They are equivariant, not invariant.
The Problem#
If we rotate the molecule by \(R\):
Energy should stay the same: \(E(R\mathbf{r}) = E(\mathbf{r})\) (invariant)
Forces should rotate: \(\mathbf{F}(R\mathbf{r}) = R\,\mathbf{F}(\mathbf{r})\) (equivariant)
Option 1: Predict energy \(E\), then compute forces via autograd: \(\mathbf{F}_i = -\nabla_{\mathbf{r}_i} E\). This is guaranteed equivariant (we used this in Lecture 22).
Option 2: Build equivariance into the network itself — this is what modern architectures like NequIP and MACE do.
# Demo: verifying rotation invariance of our GNN
torch.manual_seed(42)
gnn.eval()
# Take a test molecule
test_mol = test_data[0]
pos = torch.tensor(test_mol['positions'])
z = torch.tensor(test_mol['z'])
edge_idx = torch.tensor(test_mol['edge_index'])
# Random 3D rotation matrix
def random_rotation():
"""Generate a random 3D rotation matrix."""
# QR decomposition of random matrix gives uniform rotation
M = torch.randn(3, 3)
Q, R = torch.linalg.qr(M)
# Ensure proper rotation (det = +1)
Q = Q * torch.sign(torch.diag(R))
if torch.det(Q) < 0:
Q[:, 0] *= -1
return Q
with torch.no_grad():
E_original = gnn(z, pos, edge_idx).item()
print(f"Original energy: {E_original:.6f}")
print("\nRotated energies:")
for i in range(5):
R = random_rotation()
pos_rotated = pos @ R.T # rotate all atoms
E_rotated = gnn(z, pos_rotated, edge_idx).item()
print(f" Rotation {i+1}: {E_rotated:.6f} (diff: {abs(E_rotated - E_original):.2e})")
print("\nThe GNN prediction is rotation-invariant (by construction)!")
print("This is because we only use distances, which are rotation-invariant.")
Original energy: -0.477264
Rotated energies:
Rotation 1: -0.477264 (diff: 1.79e-07)
Rotation 2: -0.477264 (diff: 0.00e+00)
Rotation 3: -0.477264 (diff: 1.79e-07)
Rotation 4: -0.477264 (diff: 1.79e-07)
Rotation 5: -0.477264 (diff: 2.98e-07)
The GNN prediction is rotation-invariant (by construction)!
This is because we only use distances, which are rotation-invariant.
# Computing forces via autograd (equivariant by construction)
pos_grad = pos.clone().requires_grad_(True)
E = gnn(z, pos_grad, edge_idx)
E.backward()
forces = -pos_grad.grad # F = -dE/dr
print("Forces from autograd:")
for i, (elem, f) in enumerate(zip(['A', 'A', 'A', 'A'], forces)):
print(f" Atom {i}: F = [{f[0]:.4f}, {f[1]:.4f}, {f[2]:.4f}]")
# Verify equivariance: rotate, then compute forces
R = random_rotation()
pos_rot = (pos @ R.T).clone().requires_grad_(True)
E_rot = gnn(z, pos_rot, edge_idx)
E_rot.backward()
forces_rot = -pos_rot.grad
# Expected: forces_rot = R @ forces
forces_expected = forces @ R.T
error = (forces_rot - forces_expected).abs().max().item()
print(f"\nForce equivariance error: {error:.2e}")
print("Forces transform correctly under rotation!")
Forces from autograd:
Atom 0: F = [1.0334, 1.0510, 0.4401]
Atom 1: F = [0.7535, 0.1950, 0.4841]
Atom 2: F = [-0.6574, -0.1661, -0.6888]
Atom 3: F = [-1.1295, -1.0799, -0.2353]
Force equivariance error: 4.95e-06
Forces transform correctly under rotation!
VI. The Landscape of Modern GNN Architectures#
Our simple GNN is a good starting point, but research has produced many more powerful architectures:
Invariant Models (use distances only)#
Model |
Key Idea |
Paper |
|---|---|---|
SchNet (2017) |
Continuous convolutions on distances |
Schütt et al. |
DimeNet (2020) |
Adds angles between triplets of atoms |
Gasteiger et al. |
GemNet (2021) |
Adds dihedral angles (4-body interactions) |
Gasteiger et al. |
Equivariant Models (use directional information)#
Model |
Key Idea |
Paper |
|---|---|---|
PaiNN (2021) |
Equivariant message passing with vectors |
Schütt et al. |
NequIP (2022) |
E(3)-equivariant with spherical harmonics |
Batzner et al. |
MACE (2022) |
Multi-body equivariant messages |
Batatia et al. |
EquiformerV2 (2023) |
Equivariant transformer |
Liao et al. |
EOSnet (2025) |
Equivariant orbital-based many-body features (our group) |
Tao and Zhu |
The Key Progression#
Distances only → + Angles → + Directions (vectors)
(SchNet) (DimeNet) (NequIP, MACE)
2-body info → 3-body info → Many-body info
r_ij r_ij, θ_ijk Equivariant tensor products
More geometric information → better accuracy → but more computation.
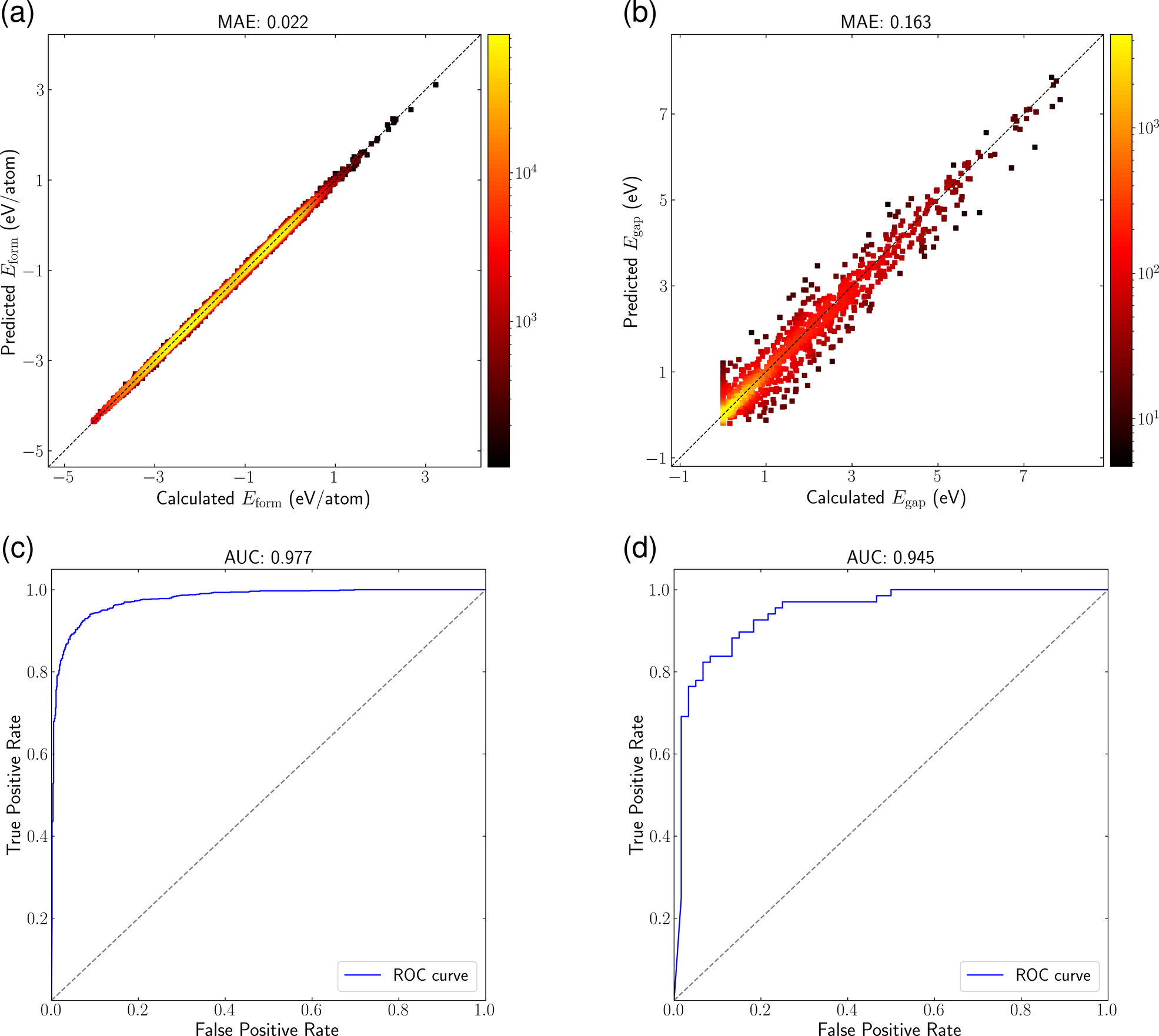
Evaluation for regression and binary classification tasks on the performance of our EOSnet model. (a) Parity plot for formation energy predictions on 131,240 data points from the Materials Project, with MAE of 0.022 eV/atom. (b) Parity plot for the prediction of electronic band gap (Eg) using 19,393 data points from the Materials Project, showing an MAE of 0.163 eV. (c) ROC curve for metal/nonmetal classification, achieving an AUC of 0.977. (d) ROC curve for dynamically stable/unstable classification on 1,335 guest-atom-substituted type-VII boron-carbide clathrates (MB6–xCx, x from 1 to 5), achieving an AUC of 0.945.
EOSnet: Rutgers-ZRG/EosNet
VII. Real-World Impact: ML Interatomic Potentials#
GNN-based interatomic potentials are now a major tool in computational materials science and chemistry:
The Speed–Accuracy Trade-off#
Accuracy ↑
| ★ Coupled Cluster (CCSD(T))
| ★ DFT
| ★ ML Potentials (GNN)
| ★ Classical Force Fields
|-------------------------------------------→ Speed
ML potentials achieve DFT-level accuracy at force-field speed:
DFT: ~hours for 100 atoms
ML potential: ~milliseconds for 100 atoms
Speedup: \(10^4\) – \(10^6\) times
Applications#
Application |
Method |
Impact |
|---|---|---|
Molecular dynamics |
MACE, NequIP |
Simulate millions of timesteps at DFT accuracy |
Drug discovery |
SchNet, DimeNet |
Screen molecular properties |
Materials design |
MEGNet, CGCNN |
Predict crystal properties |
Catalysis |
GemNet, EquiformerV2 |
Find optimal catalyst surfaces |
Protein folding |
GNNs in AlphaFold |
Predict 3D structure from sequence |
Universal ML Potentials#
Recent “foundation models” for atomic systems are trained on massive DFT datasets and work across the periodic table:
MACE-MP-0 (2023): Trained on Materials Project data, works for any element
CHGNet (2023): Includes charge information
M3GNet (2022): Universal potential for materials
These can be used as starting points and fine-tuned for specific systems.
VIII. Using Pre-trained GNNs with ASE#
In practice, you often use pre-trained GNN potentials rather than training from scratch. The ASE (Atomic Simulation Environment) library provides a convenient interface.
Here is the typical workflow (not executed in class, but important to know):
from ase import Atoms
from ase.optimize import BFGS
## Example with MACE (if installed)
## from mace.calculators import mace_mp
## Create a molecule
water = Atoms('H2O', positions=[
[0.0, 0.0, 0.0],
[0.757, 0.587, 0.0],
[-0.757, 0.587, 0.0]
])
## Attach ML calculator
## calc = mace_mp(model="medium", device="cpu")
## water.calc = calc
## Get energy and forces
## E = water.get_potential_energy()
## F = water.get_forces()
## Optimise geometry
## opt = BFGS(water)
## opt.run(fmax=0.01)
This is the same interface as DFT calculators — you can swap between ML potentials and DFT seamlessly!
IX. GNN Beyond Energy: Property Prediction#
GNNs can predict many molecular/material properties beyond energy. Let’s build a simple property predictor.
# Create a synthetic dataset of "molecules" with different atom types
# and a target property that depends on composition and geometry
np.random.seed(42)
def synthetic_property(atomic_numbers, positions):
"""A synthetic property that depends on composition and geometry.
Mimics something like a dipole moment or HOMO-LUMO gap."""
N = len(atomic_numbers)
# Composition term
comp = sum(z**0.5 for z in atomic_numbers) / N
# Geometry term (average nearest-neighbour distance)
dists = []
for i in range(N):
min_d = float('inf')
for j in range(N):
if i != j:
d = np.linalg.norm(positions[i] - positions[j])
min_d = min(min_d, d)
dists.append(min_d)
avg_nn = np.mean(dists)
# Property = f(composition, geometry) + noise
return comp * avg_nn + 0.1 * np.random.randn()
# Generate molecules with 3-6 atoms, types 1-3
mol_dataset = []
for _ in range(1500):
n_atoms = np.random.randint(3, 7)
z = np.random.randint(1, 4, size=n_atoms) # atom types 1, 2, 3
pos = np.random.randn(n_atoms, 3).astype(np.float32) * 1.2
# Check distances
min_dist = float('inf')
for i in range(n_atoms):
for j in range(i+1, n_atoms):
min_dist = min(min_dist, np.linalg.norm(pos[i] - pos[j]))
if min_dist < 0.5:
continue
prop = synthetic_property(z, pos)
edge_index = build_graph(pos, cutoff=3.5)
mol_dataset.append({
'z': z.astype(np.int64),
'positions': pos,
'edge_index': edge_index.astype(np.int64),
'energy': prop, # reuse 'energy' key
})
print(f"Generated {len(mol_dataset)} molecules")
sizes = [len(d['z']) for d in mol_dataset]
print(f"Sizes: {min(sizes)}-{max(sizes)} atoms")
print(f"Property range: [{min(d['energy'] for d in mol_dataset):.2f}, "
f"{max(d['energy'] for d in mol_dataset):.2f}]")
print("\nNote: same GNN handles molecules of DIFFERENT sizes — this is")
print("impossible with a fixed-size MLP!")
Generated 1420 molecules
Sizes: 3-6 atoms
Property range: [0.70, 7.34]
Note: same GNN handles molecules of DIFFERENT sizes — this is
impossible with a fixed-size MLP!
# Train on variable-size molecules
torch.manual_seed(42)
n_tr = int(0.8 * len(mol_dataset))
tr_mols = mol_dataset[:n_tr]
te_mols = mol_dataset[n_tr:]
# Normalise
P_mean = np.mean([d['energy'] for d in tr_mols])
P_std = np.std([d['energy'] for d in tr_mols])
for d in mol_dataset:
d['energy_norm'] = (d['energy'] - P_mean) / P_std
gnn_prop = SimpleGNN(n_elements=4, node_dim=32, edge_dim=16, n_layers=4)
optimizer = torch.optim.Adam(gnn_prop.parameters(), lr=0.002)
batch_size = 32
train_l, val_l = [], []
for epoch in range(150):
gnn_prop.train()
np.random.shuffle(tr_mols)
epoch_loss = 0
nb = 0
for i in range(0, len(tr_mols), batch_size):
batch = collate_graphs(tr_mols[i:i+batch_size])
optimizer.zero_grad()
pred = gnn_prop(batch['z'], batch['positions'],
batch['edge_index'], batch['batch'])
true = torch.tensor([d['energy_norm'] for d in tr_mols[i:i+batch_size]],
dtype=torch.float32)
loss = F.mse_loss(pred, true)
loss.backward()
optimizer.step()
epoch_loss += loss.item()
nb += 1
train_l.append(epoch_loss / nb)
gnn_prop.eval()
with torch.no_grad():
vb = collate_graphs(te_mols)
vp = gnn_prop(vb['z'], vb['positions'], vb['edge_index'], vb['batch'])
vt = torch.tensor([d['energy_norm'] for d in te_mols], dtype=torch.float32)
val_l.append(F.mse_loss(vp, vt).item())
if epoch % 30 == 0:
print(f"Epoch {epoch:3d} Train: {train_l[-1]:.5f} Val: {val_l[-1]:.5f}")
# Parity plot
gnn_prop.eval()
with torch.no_grad():
vb = collate_graphs(te_mols)
pred = gnn_prop(vb['z'], vb['positions'], vb['edge_index'], vb['batch']).numpy()
pred_real = pred * P_std + P_mean
true_real = np.array([d['energy'] for d in te_mols])
fig, ax = plt.subplots(figsize=(6, 5))
ax.scatter(true_real, pred_real, s=15, alpha=0.5)
lims = [min(true_real.min(), pred_real.min()), max(true_real.max(), pred_real.max())]
ax.plot(lims, lims, 'r--', lw=2)
ax.set_xlabel('True Property'); ax.set_ylabel('GNN Prediction')
ax.set_title(f'Variable-size molecules (R² = {r2_score(true_real, pred_real):.3f})')
ax.grid(alpha=0.3)
plt.tight_layout()
plt.show()
print("The same GNN handles 3-atom, 4-atom, 5-atom, and 6-atom molecules!")
Epoch 0 Train: 0.68437 Val: 0.52358
Epoch 30 Train: 0.07135 Val: 0.07400
Epoch 60 Train: 0.05304 Val: 0.04682
Epoch 90 Train: 0.04971 Val: 0.06432
Epoch 120 Train: 0.03480 Val: 0.07522
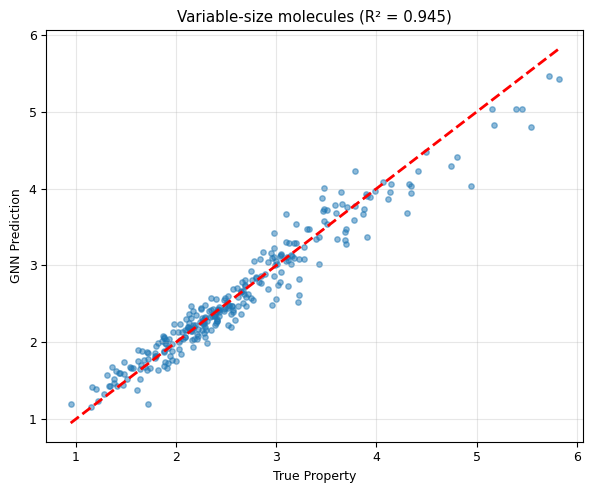
The same GNN handles 3-atom, 4-atom, 5-atom, and 6-atom molecules!
Summary#
Concept |
Key Idea |
|---|---|
Graphs |
Atoms = nodes, interactions = edges |
Message passing |
message → aggregate → update |
Radial basis functions |
Encode distances as feature vectors |
Permutation invariance |
Sum aggregation over neighbours |
Rotation invariance |
Use distances (not coordinates) |
Rotation equivariance |
Forces via autograd, or equivariant layers |
Spherical harmonics |
Basis for equivariant features |
Variable size |
Same model for any number of atoms |
The ML for Physics Pipeline#
Lecture 21: ML foundations — Fit models, classify, evaluate
Lecture 22: Neural networks — Learn functions, autograd for forces
Lecture 23: Physics-informed — Embed PDEs, discover order parameters
Lecture 24: Graph networks — Encode structure, symmetry, scalability
The big picture: ML in physics works best when we combine data with physical knowledge — symmetries, conservation laws, and the right representation of the problem.